Fabry disease & Smart4Fabry project
The Fabry disease (FD) is a lysosomal storage disorder (LSD) that currently lacks an effective treatment. Lysosomes are spherical vesicles, which contain hydrolytic enzymes found in nearly all animal cells. LSDs are caused by lysosomal dysfunctions, usually because of the deficiency of a single enzyme required for the metabolism of macromolecules such as lipids, glycoproteins and mucopolysaccharides. Fabry disease is a progressive, X-linked inherited disorder caused by deficiency or absence of the α-galactosidase A (GLA) activity, an enzyme involved in the glycosphingolipid metabolism. The substrates of GLA are glycosphingolipids, being the primary substrate the globotriaosylceramide (Gb3). Therefore, the failure of GLA activity leads to progressive intracellular accumulation of Gb3, in many cells, particularly in renal epithelial cells, endothelial cells, pericytes, vascular smooth muscle cells, cardiomyocytes, and neurons of the autonomic nervous system, leading to multisystemic clinical symptoms. First clinical signs of FD occur during childhood and, over time, microvascular lesions of the affected organs progress leading to early death. It affects mostly men but serious cases have also been reported in women.
There are currently three products authorized in the EU for the treatment of FD. Two products available in EU since 2001 for Enzymatic Replacement Therapy (ERT), Replagal (Shire Human Genetic Therapies AB) and Fabrazyme (Genzyme Europe B.V.), which have to be i.v. administered every other week. The ERT strategy is based on supplying recombinant GLA to cells, reversing several of the metabolic and pathologic abnormalities. There is a third product in the EU market since 2016, which is based on the chaperone migalastat hydrochloride (Galafold Amicus Therapeutics UK Ltd), designed to selectively and reversibly bind with high affinity to the active sites of certain mutant forms of GLA, facilitating proper protein folding and allowing for correct trafficking of the mutant enzyme. However, it is a genotype-specific treatment (only one-third to one-half of mutations may be amenable).
To date, no direct comparisons exist between Fabrazyme and Replagal but significant clinical benefits compared with placebo, however, have been demonstrated with ERT, with positive effects on the heart, kidneys, nervous system and quality of life. Of note, a stabilization of renal function was only observed at an early phase of FD.
ERT success with free GLA is limited mainly due to the instability and low efficacy of the exogenously administered therapeutic enzyme. Furthermore, some patients can develop immune responses after receiving the infused recombinant enzyme. Clinical data has confirmed that the immunological consequences of ERT may impair efficacy in some patients. Furthermore, the short elimination t1/2 of the enzyme and the need for repeated administration of large amounts of enzyme are other limitations of current ERT. In addition, GLA does not cross of the Blood Brain Barrier (BBB), which prevents the product for reducing the Gb3 deposits in the central nervous system (CNS). Moreover, it is a lifelong treatment which becomes a burden for the health system due to its extremely high cost.
Therefore, there is a need for other therapeutic strategies, which can either serve as primary or supplemental treatments. Gene and substrate reduction therapies constitute alternative therapies which are at present under investigation.
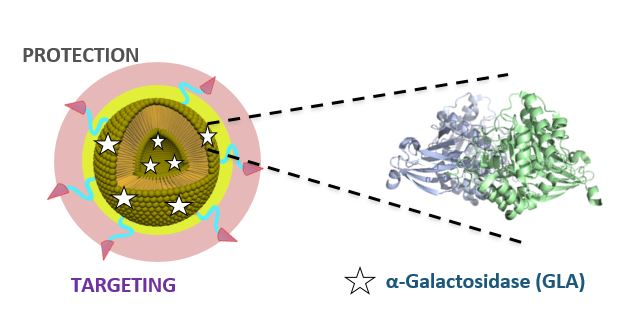
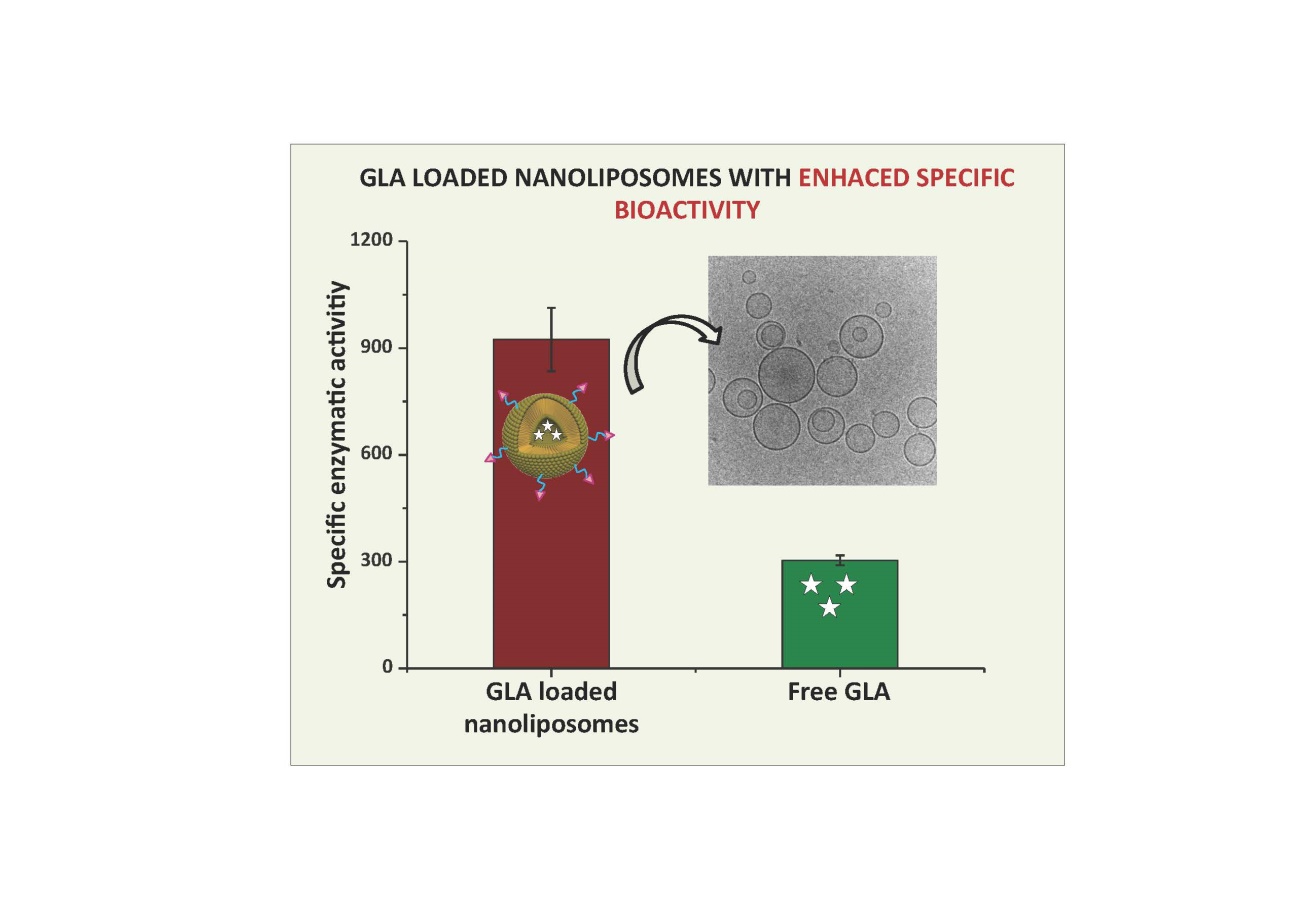
The European “Smart-4-Fabry” project aims to develop a new nanoformulation based on the encapsulation of the GLA enzyme in nanoliposomes, to improve the current ERT of FD. A Consortium formed by ten partners, including private companies and public institutions in Europe and Israel, has been granted (July 2017) with a Horizon2020 financial programme by the European Commission (H2020-NMBP-2016-2017; call for nanotechnologies, advanced materials, biotechnology and production; Proposal number: 720942-2).
The project is expecting to last for 48 months and contemplates the necessary activities to advance a nanoliposome formulation of GLA enzyme, i.e., nano-GLA, from an experimental proof of concept up to an advanced nonclinical stage of development. The S4F should complete an advanced regulatory safety and toxicology package supporting future nano-GLA clinical development in patients with FD.
To the best of S4F knowledge, there is no previous experience on the encapsulation of a GLA for treating FD patients following an ERT approach.